 LOGIN
LOGIN REGISTER
REGISTER.png)


Review Article
Therapeutic Potential for Natural Product HDAC Inhibitors in Heart Failure
Levi W Evans1,2, Samantha S Romanick1,3 and Bradley S Ferguson1*
1Department of Agriculture, Nutrition & Veterinary Sciences, University of Nevada, Reno, Nevada, USA
2Environmental Sciences & Health, University of Nevada, Reno, USA
3Cellular & Molecular Pharmacology and Physiology, University of Nevada, Reno, USA
*Corresponding author: Bradley S Ferguson, Department of Agriculture, Nutrition & Veterinary Sciences, University of Nevada, 1321 Gleneyre Ct., Reno, NV 89509, USA, Tel: +1 7757846278; E-mail: bferguson@unr.edu
Citation: Evans LW, Romanick SS, Ferguson BS (2017) Therapeutic Potential for Natural Product HDAC Inhibitors in Heart Failure. J Pharmacol Clin Trials 2017: 1-7. doi: https://doi.org/10.29199/JPCT.101011
Received Date: 31 May, 2017; Accepted Date: 22 June, 2017; Published Date: 24 July, 2017
Abstract
Histone Deacetylases (HDACs) catalyze the removal of acetyl-groups from lysine residues of a variety of proteins, but have traditionally been studied in the regulation of chromatin remodeling and gene expression. Past research demonstrated that HDAC inhibition was efficacious in pre-clinical animal models of heart failure, in which small-molecule HDAC inhibitors blocked cardiac remodeling (e.g. hypertrophy) and improved cardiac systolic function. Many bioactive compounds found in edible plants have recently been shown to inhibit HDAC activity that mechanistically impacted inflammation, cardiac hypertrophy, and cardiac fibrosis. This new area of research has given rise to the study of nutrient-epigenetic-gene interactions, nutri-epigenetics in the regulation of human health and disease. Thus, bioactive compounds that function as HDAC inhibitors offer promising therapeutic strategies for the prevention and treatment of cardiac disease and will be discussed in this review.
Keywords: Acetylation; Bioactive compounds; Dietary HDAC Inhibitors HDACs; Histone Deacetylases
Introduction
Cardiovascular disease encompasses a range of conditions that impact the heart, which includes hypertension and myocardial infarction. In response to stress, such as hypertension, the myocardium undergoes remodeling that is characterized by muscle cell hypertrophy, apoptosis and fibrosis. Cardiac remodeling often results in impaired cardiac function and ultimately Heart Failure (HF) [1].
Approximately 5.7 million US adults (>20 yrs) have HF, with projections estimating a 46% increase in prevalence rates by 2030 resulting in > 8 million adults with HF [2]. This increase is due, in part, to the growing number of elderly adults in the US, as aging is an underlying risk factor for HF. Moreover, with this increase in HF prevalence, direct medical costs are projected to rise from $21 billion to $53 billion, annually, while indirect medical costs are projected to increase from $31 billion to $70 billion [3]. While HF survival improved from 1979 to 2000, due to improvements in standards of care that include treatment with β-blockers, angiotensin receptor blockers and Angiotensin Converting Enzyme inhibitors (ACEi), five-year mortality rates remain high at approximately 50%, with 1-year mortality rates for Medicare beneficiaries approximating 29.6% [2]. High mortality rates combined with increased medical costs highlight the need for improved therapeutic approaches capable of managing and/or preventing this disease [4].
Current HF drugs including β-blockers and ACEi inhibit signaling pathways stimulated by cell surface receptors to prevent cardiac pump dysfunction [5]. However, redundant signaling pathways have been implicated in pathological cardiac remodeling and failure, suggesting that drugs designed to target shared downstream mediators of these signaling pathways would be more efficacious for the treatment of HF.
Epigenetics refers to global changes in gene expression that does not alter the DNA sequence. It has been postulated that drugs designed to alter the epigenome have the potential to inhibit many shared downstream targets of pathological signaling. Indeed, animal studies have elucidated important functional roles for Histone Deacetylases (HDACs) in the heart [4,6-11]. HDACs have commonly been characterized in the regulation of nucleosomal DNA where they remove acetyl groups from lysine residues on histone tails altering chromatin structure in a manner that confers transcriptional repression. Conversely, acetylation of histone proteins on lysine residues by Histone Acetyltransferases (HATs) promotes transcriptional activation [4,12-14] (Figure 1).
Histone Acetyl Transferases (HATS) acetylate lysine residues on histone tails resulting in transcriptional activation. Conversely, Histone Deacetylases (HDACs) deacetylate lysine residues resulting in transcriptional repression. HDAC inhibitors (HDACi) inhibit HDACs from removing acetyl marks from lysine residues and thus alter gene expression. |
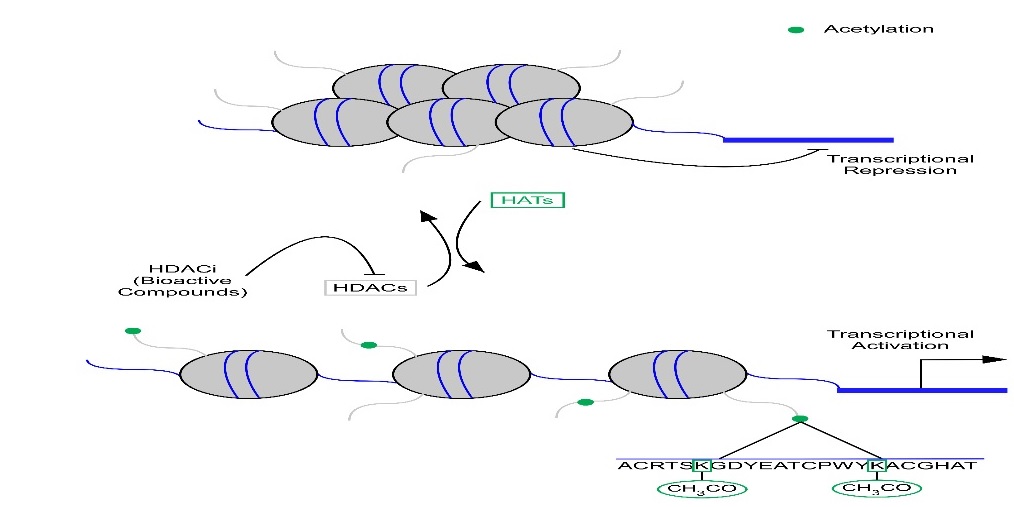
Eighteen mammalian HDACs have currently been identified and have been grouped into four distinct classes. Classes I, II, and IV are comprised of zinc dependent HDACs, where zinc is required for deacetylase activity. Class III is comprised of the sirtuin family. Sirtuins are Nicotinamide Adenine Dinucleotide (NAD+) dependent, where NAD+ is a required cofactor for deacetylase activity [13,14].
Class I is comprised of HDACs 1, 2, 3, and 8. Class II is subdivided into class IIa and class IIb, in which class IIa is comprised of HDACs 4, 5, 7, and 9 and class IIb comprised of HDACs 6 and 10. Class III is comprised of SIRTs 1 - 7. Class IV is comprised of HDAC 11 (Figure 2).
There are eighteen mammalian HDACS, which are divided into four different classes. Class , I, IIa, IIb, & IVHDACs are zinc dependent HDACs, while class IIIHDACs, also known as sirtuins, are Nicotinamide Adenine Dinucleotide (NAD+) dependent. |

Studies have shown efficacy of zinc dependent HDAC inhibition in heart failure [4,6-11]; therefore, this review will focus on zinc dependent HDACs as efficacious therapy for the heart.
There is a current shift in today’s population in which natural compounds are preferred for use in medicine over synthetically produced medications. This has driven an increased demand for natural compound discovery as potential therapeutics [15,16]. Indeed, synthetic HDAC inhibitors are currently approved as well as under clinical trials for the treatment of various cancers. However, most of these synthetic inhibitors have shown adverse effects and toxicities and are also cost prohibitive [17]. Together, this has led to recent interest in bioactive food compounds that regulate epigenetic marks in the control of gene expression and human health and disease. This review will focus on bioactive compound HDAC inhibitors and their potential as therapeutics for HF.
HDACs and the heart
Early roles for HDACs in the heart involved findings that class IIa HDACs interact with Myocyte Enhancer Factor-2 (MEF2) transcription factor family members. MEF2 is upregulated in response to cardiac hypertrophy and its expression leads to dilated cardiomyopathy [18]. Class IIa HDACs inhibit myocyte hypertrophy in vitro and in vivo, as they bind and repress MEF2 transcriptional activity, downregulating cardiac hypertrophic genes [19]. Furthermore, deletion of class IIa HDACs (HDACs 9 and 5) promoted pathological cardiac hypertrophy due to increased MEF2 transcriptional activity [20,21].
Later studies highlighted class I HDACs as detrimental to heart health. Global deletion of HDACs 1, 2, or 3 results in embryonic or perinatal lethality in rodents, while cardiac-specific deletion of HDACs 1, 2 or 3 contributed to cardiac hypertrophy and dysfunction as well as metabolic distress [22-24]. While these studies focused on loss-of HDAC function, class I HDAC activation is not necessarily advantageous in the setting of the heart. For example, activation of HDAC 2 has been shown to promote cardiac myocyte hypertrophy [25]. This would suggest that class I HDAC inhibition, but not deletion, may be efficacious in the heart.
Less is known about the class IIb HDACs in the heart. Most studies have focused on HDAC6, where it has been reported that HDAC6 activity is increased in animal and human models of hypertension [26]. HDAC6 is a cytosolic deacetylase that has been shown to deacetylate microtubules. Microtubule deacetylation impacts tubulin polymerization that has been associated with cardiac fibrosis. Genetic deletion of HDAC6 protects the heart from systolic dysfunction, in part through regulation of sarcomeric protein deacetylation and improved cardiac contractility [27]. To date, nothing is known about the other class IIb HDAC, HDAC 10 or the class IV HDAC, HDAC 11 in the heart.
Given the historical observations about class IIa HDACs and the detrimental actions for class I HDAC deletion in the heart, it was postulated that treatment with HDAC inhibitors would promote pathological cardiac remodeling. However, studies have since shown that HDAC inhibitor treatment is efficacious in pre-clinical models of HF [4,6-11]. These paradoxical findings likely reflect three important observations: 1) HDAC inhibitors possess weak actions against the class IIa HDACs and therefore do not promote cardiac remodeling via MEF2 transcriptional activation; 2) little evidence supports deacetylase activity for class IIa HDACs in vivo, with evidence demonstrating that class IIa HDACs regulate MEF2 transcriptional activity via binding interactions; and 3) inhibition of class I HDAC deacetylase activity is cardiac protective as opposed to genetic deletion, which eliminates other HDAC functions within the cell (e.g., protein scaffolding).
HDAC inhibitors
In 2006, Suberoylanilide Hydroxamic Acid (SAHA), also known as Vorinostat, was the first FDA approved HDACi to be used for the treatment of T-cell lymphoma [28]. More recently, Romidepsin (FK-228) was approved to also treat T-cell lymphoma [29]. Valproic Acid, another HDACi is also currently available for human use; initially used to treat epilepsy, it has since become prominent for other neurological-related ailments [30]. Because of this, medicinal chemistry efforts have pushed for the development of not only broad spectrum but also class-selective and isoform-specific HDAC inhibitors.
Non-Sirtuin HDACs contain a zinc-ion domain for catalyzing activity. The classical HDAC inhibitor pharmacophore consists of a three-part structure that contains a zinc-binding motif capable of binding to the active site, a surface recognition domain that interacts with residues near the active site, as well as a hydrocarbon linker that connects the motifs to the domain. Historically, HDACi have been classified into four groups: hydroxamic acids, short-chain fatty acids, benzamides, and cyclic peptides [13,14,31]. However, ortho-aminoanilide HDACi were recently discovered [32] and imply further undiscovered HDACi groups. Potencies and HDAC class selectivity differ with these classes [14].
Hydoxamic Acid HDACi, such as Trichostatin A and SAHA, are considered pan-HDACi and possess strong zinc-chelating properties, which yield potent, low nanomolar, HDACi. Conversely, short-chain fatty acids, such as Valproic Acid and sodium butyrate, tend to be weak HDACi (millimolar). Their fatty acid physiochemical properties, which allow for advantageous uptake and transportation, give promising efficacy, however come with limitations (e.g., nonspecific biochemical targets). Benzamide HDACi, such as MS-275, contain a benzene ring as the hydrocarbon linker and an amide motif and are characteristically selective for class I HDACs (HDACs 1, 2 and 3) [31]. Ortho-aminoanilide is similar in structure to the benzamide HDACi. Cyclic peptide HDACi, such as Apicidin and Romidepsin, are often highly potent and incorporate alkyl-linking motifs as well as various zinc-binding groups. Similar to benzamide HDAC inhibitors, cyclic peptide HDAC inhibitors tend to be selective for class I HDACs. Other HDACi are very selective towards specific HDACs, and are generally referred to as isoform-selective HDACi; Tubacin, for example is considered to have limited selectivity for HDAC6/class IIb [33].
HDAC inhibitors and heart failure
In vivo studies have demonstrated that pan- and isoform-selective HDAC inhibitors block and potentially reverse cardiac remodeling in the heart. These studies have been reviewed extensively [4,6,7,11,34,35]. Treatment with pan-HDAC inhibitors such as TSA, for instance, have been shown to block and even reverse cardiac hypertrophy and systolic dysfunction in rodent models of aortic constriction. Cardiac myocytes and transgenic mice experience significant reductions in cardiac hypertrophy after Trichostatin A treatment [36-38].
The pan-HDACi, MPT0E014, substantially represses expression of the pathogenic HF-indicators, transforming growth factor beta (TGF-β), calmodulin-dependent protein kinase (CaMKIIδ) and angiotensin II type I receptor in HF-induced rats, which led reduction in left ventricular wall thickening as well as increased ejection fraction and fraction shortening [39]. MPT0E014 has also been shown to reduce inflammation and Peroxisome Proliferator-Activated Receptor (PPAR) dysregulation in response to diabetic cardiomyopathy [40] of significance, SAHA, an FDA approved pan-HDACi, decreased infarct size and improved systolic heart function in a rabbit model of Ischemia-Reperfusion (I/R) injury; this is important as delivery of SAHA before or during reperfusion was efficacious [8]. SAHA, has also been shown to inhibit systolic blood pressure, collagen formation, cardiac stiffness, left ventricle hypertrophy, action potential duration, and vascular dysfunction based off relaxation and contraction force [41].
More recently, we reported that class I HDACs regulated cardiac hypertrophy and fibrosis [42,43]. In these reports, we showed that class I-selective HDAC inhibition blocked cardiac myocyte hypertrophy and angiotensin II-dependent fibrosis. Indeed, others have shown that the class I HDAC inhibitor, Mocetinostat, has had much success in alleviating and improving HF-related events, both in vivo and in vitro [44,45]. Similarly, treatment with class I-selective short chain fatty acid HDAC inhibitors, Tributyrin or Valproic Acid, inhibited cardiac hypertrophy and fibrosis in a rat infarct model [46]. Short chain fatty acid HDAC inhibitor specificity has been questioned, as these compounds regulate many non-HDAC mediated mechanisms and are involved in a number of biochemical pathways [30]. Nonetheless, short chain fatty acid HDAC inhibitors have been used successfully in humans for other ailments and thus have the potential to treat human HF.
Lastly, studies examining class IIb HDACs in the heart have focused on HDAC6. Early observations regarding HDAC6 inhibition demonstrated a role for HDAC6 in the regulation of myofibril contractility, in which animals treated with HDAC6 inhibitors were protected from systolic dysfunction in response to angiotensin II [27]. Others have shown that inhibition of HDAC6 improved cardiac function in a mouse model of cardiac proteotoxicity, in part, via regulation of autophagy-mediated degradation [47].
Bioactive HDACi
HDACi can be synthetically and naturally produced. Some of the more potent and commonly used HDACi have been isolated from natural plants, herbs, and food stuffs. The benefits from consuming fruits, vegetables, and whole grains have been well-studied and well-established [48]; focus has now turned to their underlying mechanisms. HDACi, and other compounds, derived from plants, herbs, and foods stuffs are known to be bioactive compounds [15]. Bioactive HDACi as well as their descriptions and specifications can be found in some detail [49,50]. Cyclic peptide and short-chain fatty acid HDACi are the more familiar types of bioactive HDACi. Sodium butyrate, for example, is a short-chain fatty acid that is produced by gastrointestinal bacteria via fermentation of fibrous foods, i.e., fruits, vegetables, and whole grains. As an HDACi, it is selective towards HDACs 1 and 3, class I HDACs associated with HF. The previously described Valproic Acid is also a short-chain fatty acid HDACi [30]. Treatment with sodium butyrate or valproic acid inhibited cardiac hypertrophy and fibrosis and increased systolic function in pre-clinical rodent models of heart failure [46,51].
Other bioactive compounds from foods have been identified that regulate HDAC activity, including curcumin and resveratrol [50,52,53]. Resveratrol has been shown to regulate Sirtuins and has been extensively reviewed. Curcumin has been shown to regulate both Histone Acetyl Transferases (HATs) as well as HDACs [54,55]. For instance, curcumin was reported to inhibit p300 HAT activity thus inhibiting GATA4 transcriptional activation and hypertrophic gene expression; cardiac function was subsequently improved in a rodent model of hypertension [54]. Sulforaphane is an HDACi found in broccoli and has been shown to inhibit HDAC activity and increase histone H3 and H4 acetylation in cells, mice and man [50,53,56,57].
While most of these studies have focused on anti-carcinogenic actions for sulforaphane, other reports have shown benefit for sulforaphane treatment in models of diabetic cardiomyopathy. More recently, grape seed procyanidin extract was found to significantly inhibit HDAC activity in rat liver, concomitant with reductions in serum triglycerides [58]. Indeed, the field of nutrigenomics and nutri-epigenetics has rapidly advanced in the last decade leading to increased reports demonstrating the potential impact for food compounds in the regulation of epigenetic marks and gene expression [49,50].
Most of these studies however, have focused on bioactive food compounds in the treatment or prevention of cancer [49,50,59]; limited studies have examined dietary HDAC inhibitors in the regulation of cardiovascular disease.
Recently, we screened several isolated bioactive compounds (131 compounds) found in plants, herbs, and food stuffs as potential inhibitors of HDAC activity in the bovine heart [60]. HDACs were specific to the non-sirtuin, zinc-dependent HDACs- class I, IIa, and IIb. Of the 131 screened compounds, we reported that eighteen inhibited HDAC activity; these eighteen compounds are described in (Table 1).
Table 1: Dietary HDAC inhibitors.The table represents the eighteen compounds isolated and assessed for HDAC activity inhibition for class I, IIa and IIb. |
HDAC inhibitor therapy is efficacious and prevalent in pharmacology (e.g., Vorinostat, Romidepsin, and Valproic Acid); however, most studies have focused on HDAC inhibitor therapy in humans for the treatment of cancer and neurodegenerative-related disorders such as epilepsy and depression [61]. While HDAC inhibitors appear efficacious in pre-clinical models of HF, future studies regarding the role for pan-, class- and isoform-selective HDAC inhibitors on human pathology and metabolism are warranted and require further investigation. Vorinostat (SAHA) poses a potential option for the treatment of human HF due, in part, to its therapeutic efficacy in rabbit I/R, which established a large animal proof-of-concept and set the stage for future clinical trials in humans [8]. In addition, SAHA is currently approved by the FDA for treatment of cutaneous T-cell lymphoma [41].
While drugs like SAHA require FDA approval, bioactive food compounds such as sulforaphane have less FDA oversight due in part to the DSHEA act of 1994. This argues that bioactive food compound HDAC inhibitors have the potential to see human studies more readily than current HDAC inhibitor therapies examined. Sulforaphane is an HDACi found in broccoli and has been shown to increase circulating acetylated histones, 3 and 4, in healthy humans [53]. These studies were performed acutely (< 24 hrs) and no other variables were measured including heart function. However, these data argue that sulforaphane passed the intestinal epithelial barrier to inhibit HDAC activity in the blood. Future studies examining sulforaphane supplementation or broccoli feeding in HF patients would be of particular interest, in which blood diagnostics examining circulating Atrial Naturetic Factor (ANF), a classical HF biomarker, as well as echocardiography for cardiac wall thickness and function could be employed.
Conclusion
The effect nutrition has on epigenetics has been termed nutri-epigenetics [62]. Nutri-epigenetics has been extensively studied in the cancer field. Because of this, bioactive HDAC inhibitors in fruits, vegetables, herbs, and food stuffs are being delineated at an exponential rate. The potential for newly found compounds is limitless as there are many types including polyphenols and flavonoids in a variety of plants, herbs, and food stuffs. Their possible application in overall pathogenesis, including HF, deserves further investigation in the scope of epigenetic modification as well as other underlying mechanisms. Lastly, bioactive food compound HDAC inhibitors offer exciting opportunities for drug development as well as prevention and treatment strategies for HF.
Acknowledgements
This work is supported by the USDA National Institute of Food and Agriculture (Hatch-NEV00727) to B.S.F. Core facilities used for Research reported in this publication was supported by National Institute of General Medical Sciences of the National Institutes of Health under grant number P20 GM103554.
Conflict of Interest
The authors declare no conflicts of interest.
References
- Aurigemma GP, de Simone G, Fitzgibbons TP (2013) Cardiac remodeling in obesity. Circ Cardiovasc Imaging 6: 142-152.
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, et al. (2015) Heart disease and stroke statistics--2015 update: a report from the American Heart Association. Circulation 131: 29-322.
- Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, et al. (2017) Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation 135: 146-603.
- McKinsey TA (2012) Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol 52: 303-319.
- Mann DL, Bristow MR (2005) Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation 111: 2837-2849.
- McKinsey TA (2011) Isoform-selective HDAC inhibitors: closing in on translational medicine for the heart. J Mol Cell Cardiol 51: 491-496.
- Ferguson BS, McKinsey TA (2015) Non-Sirtuin histone deacetylases in the control of cardiac aging. J Mol Cell Cardiol 83: 14-20.
- Xie M, Kong Y, Tan W, May H, Battiprolu PK, et al. (2014) Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation 129: 1139-1151.
- Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, et al. (2006) Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation 113: 2579-2588.
- Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, et al. (2011) Histone Deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA 108: 4123-4128.
- Berry JM, Cao DJ, Rothermel BA, Hill JA (2008) Histone deacetylase inhibition in the treatment of heart disease. Expert Opin Drug Saf 7: 53-67.
- Yang XJ, Seto E (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26: 5310-5318.
- Gregoretti IV, Lee YM, Goodson HV (2004) Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol 338: 17-31.
- Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, et al. (2010) Chemical phylogenetics of histone deacetylases. Nat Chem Biol 6: 238-243.
- Rein MJ, Renouf M, Cruz-Hernandez C, Actis-Goretta L, Thakkar SK, et al. (2013) Bioavailability of bioactive food compounds: a challenging journey to bioefficacy. Br J Clin Pharmacol 75: 588-602.
- Suleria HA, Osborne S, Masci P, Gobe G (2015) Marine-based nutraceuticals: an innovative trend in the food and supplement industries. Mar Drugs 13: 6336-6351.
- Shukla S, Meeran SM, Katiyar SK (2014) Epigenetic regulation by selected dietary phytochemicals in cancer chemoprevention. Cancer Lett 355: 9-17.
- Xu J, Gong NL, Bodi I, Aronow BJ, Backx PH, et al. (2006) Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J Biol Chem 281: 9152-9162.
- McKinsey TA (2007) Derepression of pathological cardiac genes by members of the CaM kinase superfamily. Cardiovasc Res 73: 667-677.
- Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, et al. (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110: 479-488.
- Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, et al. (2004) Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 24: 8467-8476.
- Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, et al. (2008) Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest 118: 3588-3597.
- Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J, et al. (2007) Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth and contractility. Genes Dev 21: 1790-1802.
- Sun Z, Singh N, Mullican SE, Everett LJ, Li L, et al. (2011) Diet-induced lethality due to deletion of the Hdac3 gene in heart and skeletal muscle. J Biol Chem 286: 33301-33309.
- Eom GH, Nam YS, Oh JG, Choe N, Min HK, et al. (2014) Regulation of acetylation of histone deacetylase 2 by p300/CBP-associated factor/histone deacetylase 5 in the development of cardiac hypertrophy. Circ Res 114: 1133-1143.
- Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, et al. (2011) Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J Mol Cell Cardiol 51: 41-50.
- Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, et al. (2014) HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol 307: 252-258.
- Marks PA, Breslow R (2007) Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol 25: 84-90.
- Iyer SP, Foss FF (2015) Romidepsin for the treatment of peripheral T-Cell lymphoma. Oncologist 20: 1084-1091.
- Terbach N, Williams RS (2009) Structure-function studies for the panacea, valproic acid. Biochem Soc Trans 37: 1126-1132.
- Wagner FF, Weiwer M, Lewis MC, Holson EB (2013) Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics 10: 589-604.
- Wagner FF, Weiwer M, Steinbacher S, Schomburg A, Reinemer P, et al. (2016) Kinetic and structural insights into the binding of Histone Deacetylase 1 and 2 (HDAC1, 2) inhibitors. Bioorg Med Chem 24: 4008-4015.
- Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL (2003) Domain-selective small-molecule inhibitor of Histone Deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA 100: 4389-4394.
- Stratton MS, McKinsey TA (2016) Epigenetic regulation of cardiac fibrosis. J Mol Cell Cardiol 92: 206-213.
- Schuetze KB, McKinsey TA, Long CS (2014) Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs. J Mol Cell Cardiol 70: 100-107.
- Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, et al. (2003) Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem 278: 28930-28937.
- Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, et al. (2006) Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation 113: 51-59.
- Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, et al. (2003) Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest 112: 863-871.
- Kao YH, Liou JP, Chung CC, Lien GS, Kuo CC, et al. (2013) Histone deacetylase inhibition improved cardiac functions with direct antifibrotic activity in heart failure. Int J Cardiol 168: 4178-4183.
- Lee TI, Kao YH, Tsai WC, Chung CC, Chen YC, et al. (2016) HDAC inhibition modulates cardiac PPARs and fatty acid metabolism in diabetic cardiomyopathy. PPAR Res 2016: 5938740.
- Iyer A, Fenning A, Lim J, Le GT, Reid RC, et al. (2010) Antifibrotic activity of an inhibitor of histone deacetylases in DOCA-salt hypertensive rats. Br J Pharmacol 159: 1408-1417.
- Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, et al. (2014) Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol 67: 112-125.
- Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, et al. (2013) Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad Sci USA 110: 9806-9811.
- Nural-Guvener HF, Zakharova L, Nimlos J, Popovic S, Mastroeni D, et al. (2014) HDAC class I inhibitor, mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 7: 10.
- Nural-Guvener H, Zakharova L, Feehery L, Sljukic S, Gaballa M (2015) Anti-fibrotic effects of class I HDAC inhibitor, mocetinostat is associated with IL-6/Stat3 signaling in ischemic heart failure. Int J Mol Sci 16: 11482-11499.
- Lee TM, Lin MS, Chang NC (2007) Inhibition of histone deacetylase on ventricular remodeling in infarcted rats. Am J Physiol Heart Circ Physiol 293: 968-977.
- McLendon PM, Ferguson BS, Osinska H, Bhuiyan MS, James J, et al. (2014) Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy. Proc Natl Acad Sci USA 111: 5178-5186.
- Liu RH (2013) Health-promoting components of fruits and vegetables in the diet. Adv Nutr 4: 384-392.
- Bassett SA, Barnett MP (2014) The role of dietary Histone Deacetylases (HDACs) inhibitors in health and disease. Nutrients 6: 4273-4301.
- Kim E, Bisson WH, Lohr CV, Williams DE, Ho E, et al. (2016) Histone and non-histone targets of dietary deacetylase inhibitors. Curr Top Med Chem 16: 714-731.
- Chen Y, Du J, Zhao YT, Zhang L, Lv G, et al. (2015) Histone Deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc Diabetol 14: 99.
- Chen T, Li J, Liu J, Li N, Wang S, et al. (2015) Activation of SIRT3 by resveratrol ameliorates cardiac fibrosis and improves cardiac function via the TGF-β/Smad3 pathway. Am J Physiol Heart Circ Physiol 308: 424-434.
- Myzak MC, Ho E, Dashwood RH (2006) Dietary agents as histone deacetylase inhibitors. Mol Carcinog 45: 443-446.
- Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, et al. (2008) The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest 118: 868-878.
- Wang SH, Lin PY, Chiu YC, Huang JS, Kuo YT, et al. (2015) Curcumin-mediated HDAC inhibition suppresses the DNA damage response and contributes to increased DNA damage sensitivity. PLoS One 10: 0134110.
- Dashwood RH, Ho E (2007) Dietary histone deacetylase inhibitors: from cells to mice to man. Semin Cancer Biol 17: 363-369.
- Dashwood RH (2007) Frontiers in polyphenols and cancer prevention. J Nutr 137: 267-269.
- Downing LE, Ferguson BS, Rodriguez K, Ricketts ML (2016) A grape seed procyanidin extract inhibits HDAC activity leading to increased Pparα phosphorylation and target gene expression. Mol Nutr Food Res 61: DOI: 10.1002/mnfr.201600347.
- Imran M, Ullah A, Saeed F, Nadeem M, Arshad MU, et al. (2016) Cucurmin; anticancer & antitumor perspectives: a comprehensive review. Crit Rev Food Sci Nutr 22: 1-23.
- Godoy LD, Lucas JE, Bender AJ, Romanick SS, Ferguson BS (2017) Targeting the epigenome: screening bioactive compounds that regulate histone deacetylase activity. Mol Nutr Food Res 61: DOI: 10.1002/mnfr.201600744.
- Lockwood LE, Su S, Youssef NA (2015) The role of epigenetics in depression and suicide: a platform for gene-environment interactions. Psychiatry Res 228: 235-242.
- Remely M, Lovrecic L, de la Garza AL, Migliore L, Peterlin B, et al. (2015) Therapeutic perspectives of epigenetically active nutrients. Br J Pharmacol 172: 2756-2768.